
Distinguishing Competitive From Non-Competitive Inhibition Effects
You're staring at a raw data sheet from a kinetic assay. The numbers look fine, but something feels off. The Vmax is dropping, but the Km seems to be doing its own weird dance. Is this competitive inhibition? Or is it the non-competitive kind? Honestly? Most textbooks make this sound like a clean, binary choice. It's not. I've spent over a decade in the lab watching brilliant scientists misdiagnose their inhibition effects because they relied on a single graph or an outdated rule of thumb. Let's fix that. Today, I'm going to walk you through the gritty, real-world mechanics of distinguishing competitive from non-competitive inhibition effects, complete with the messy edge cases that textbooks conveniently ignore.
How the Mechanics Actually Work Under the Hood
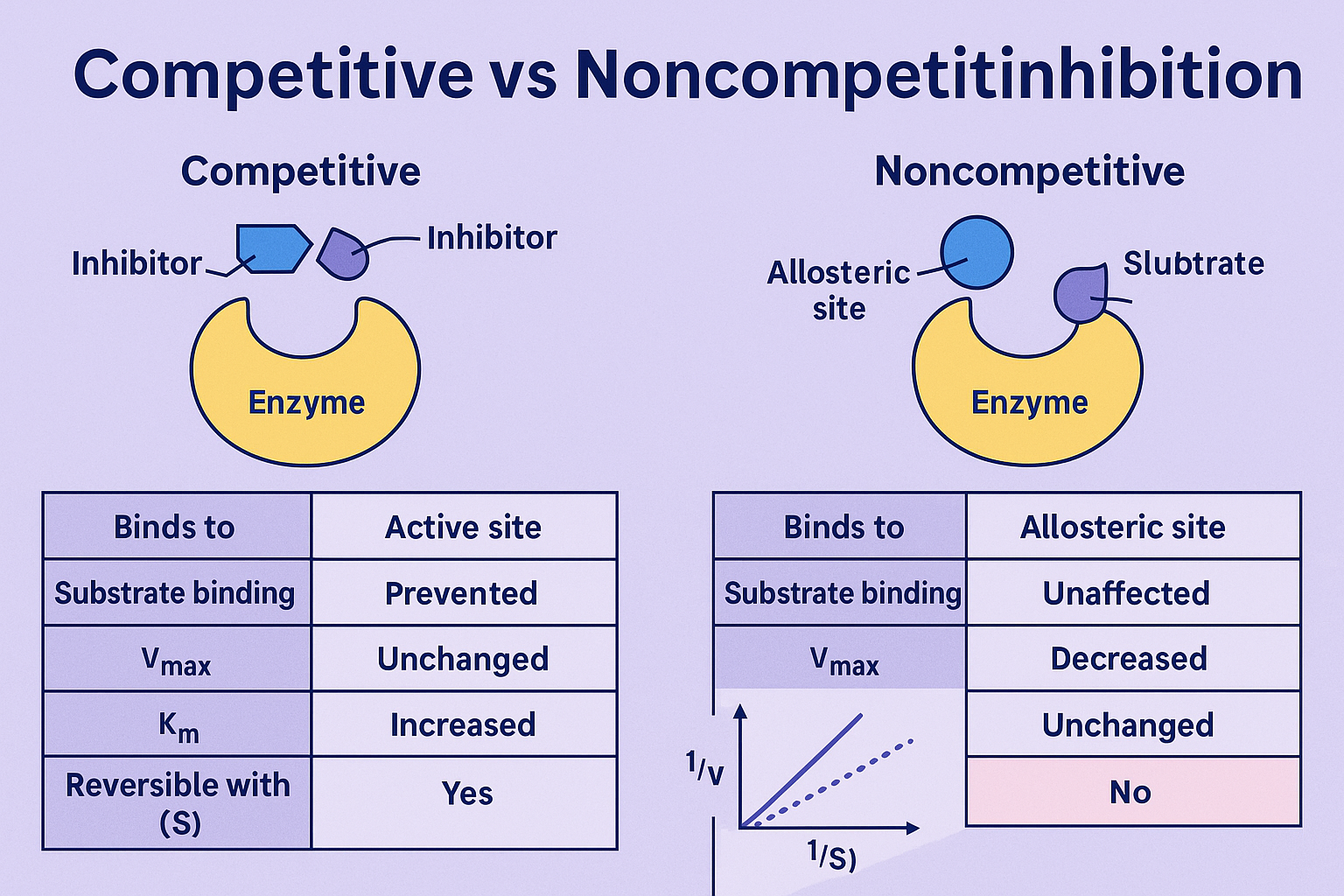
The real difference isn't just about where the inhibitor binds. It's about what happens to the enzyme's ability to function when the substrate shows up. Think of it like a lock and key analogy, but imagine someone has either stuffed gum into the keyhole or superglued the lock's internal mechanism. Both ruin your day, but the fix is completely different. That's the core of competitive inhibition versus non-competitive inhibition. The inhibitor in a competitive scenario directly competes for the active site. The non-competitive inhibitor? It binds elsewhere, often causing a conformational change that cripples the enzyme regardless of substrate concentration.
Here's the part that trips up most junior researchers: you cannot distinguish these effects by looking at the inhibition percentage alone. I've seen papers claim "mixed inhibition" based on a single inhibitor concentration, and it drives me crazy. You need a full kinetic profile. Seriously, don't cut corners here. The inhibition effects on the Michaelis constant (Km) and the maximum velocity (Vmax) are the only reliable fingerprints. A competitive inhibitor increases the apparent Km while leaving Vmax untouched. A pure non-competitive inhibitor decreases Vmax while leaving the apparent Km unchanged. That's the textbook truth, but real enzymes are rarely that polite.
Let me give you a concrete example from my work on a cytochrome P450 isoform. We had a compound that showed a 40% drop in activity. At first glance, it looked like non-competitive inhibition. But when I ran the full substrate saturation curve, the Vmax was unchanged. The Km just shifted right. That was a classic competitive profile. The compound was binding reversibly to the active site, and increasing the substrate concentration completely outcompeted it. If I had stopped at the single-point screen, I would have misclassified the entire mechanism. That's why distinguishing competitive from non-competitive inhibition effects demands a disciplined approach to data collection.
The Competitive Scenario: Substrate Overload as a Rescue Strategy
So what does a real competitive binding event look like in the raw data? You'll see the reaction velocity at low substrate concentrations plummet. But crank up the substrate concentration high enough—and I mean really high, often 10 to 20 times the Km—and the reaction recovers to nearly the uninhibited rate. That's the hallmark. The inhibitor and the substrate are fighting for the same real estate. If you flood the system with substrate, you statistically push the inhibitor out. This is why drug developers love competitive inhibition in certain contexts; it's theoretically surmountable.
But here's the nuance that textbooks skip: not all competitive inhibitors bind to the exact same subsite. Some bind near the active site and create a steric hindrance that prevents substrate entry. Others bind directly to the catalytic residues. Both are classified as competitive inhibition because they are mutually exclusive with substrate binding. However, the strength of the inhibition (the Ki value) can vary wildly based on the ionic strength of your buffer, the pH, and even the temperature. I once spent three weeks chasing a Ki that shifted by an order of magnitude simply because I was using a different lot of Tris buffer. Don't laugh. It happens.
One practical trick for distinguishing competitive from non-competitive inhibition effects in the lab: run a Lineweaver-Burk plot. I know, I know, some statisticians hate this graph because it distorts error. But for a quick visual diagnosis, it's unbeatable. A competitive inhibitor gives you lines that intersect on the y-axis. The Vmax stays constant, the slope changes. If your lines intersect on the x-axis or show no intersection at all? You're probably looking at non-competitive or mixed inhibition. Just remember: the graph is a tool, not a deity. Always confirm with a secondary analysis like a Hanes-Woolf plot or a direct fit using nonlinear regression.
The Non-Competitive Scenario: When More Substrate Doesn't Help
Now we get to the frustrating stuff. Non-competitive inhibition is the inhibitor that doesn't care how much substrate you throw at it. The Vmax drops, and it stays dropped. Period. This happens because the inhibitor binds to a site distinct from the active site, usually an allosteric site, and it reduces the enzyme's catalytic turnover rate (kcat). The enzyme is still physically capable of binding the substrate, but it's become sluggish. Imagine a factory worker who can still grab parts off the conveyor belt, but her hands are suddenly weighed down with concrete blocks. She's not blocked from the work; she's just terrible at doing it now.
The critical thing to understand is that non-competitive inhibition effects are often more dangerous in a biological context. Why? Because you cannot overcome them with a simple substrate flood. If a drug candidate shows this behavior against a human enzyme, you might be looking at a toxicity red flag. I've seen compounds get killed in late-stage development because they were non-competitive inhibitors of a CYP3A4 isoform, leading to unpredictable drug-drug interactions. The Km stays the same, the Vmax drops, and the patient's clearance rate goes haywire. It's a mess.
But here's where it gets really interesting: some enzymes exhibit a phenomenon called "partial non-competitive inhibition." The inhibitor binds, but it doesn't completely inactivate the enzyme. It just reduces the reaction velocity to a lower, but nonzero, plateau. This is extremely common in multi-subunit enzymes like hexokinase or certain dehydrogenases. If you only fit your data to a pure Michaelis-Menten model, you'll get a terrible fit and confusing parameter estimates. I always recommend using an inhibition model with an alpha term (the factor by which the inhibitor changes the binding affinity and catalytic rate) when distinguishing competitive from non-competitive inhibition effects. It adds complexity but saves you from publishing garbage data.
Practical Methods for Making the Call in the Lab
Enough theory. Let's talk about the hands-on workflow that I teach my new hires on day one. Distinguishing competitive from non-competitive inhibition effects starts with a proper experimental design, not with fancy software. You need at least five different substrate concentrations spanning a range from 0.2x Km to 5x Km. And you need at least four different inhibitor concentrations, including zero. Yes, this is a lot of pipetting. Yes, it's worth it. If you run a two-point screening and try to guess the mechanism, you are rolling dice with your career.
Once you have the data, fit it globally. Do not fit each curve separately and then compare the parameters. That introduces statistical noise. Use software like GraphPad Prism or a Python-based fitting routine that simultaneously fits all curves to both a competitive model and a non-competitive model. Compare the Akaike Information Criterion (AIC) scores. If the AIC for the competitive model is more than 7 points lower, you have your answer. If they are within 2 points of each other, you might be dealing with mixed inhibition or a more complex inhibition effect that requires a sequential model.
Another trick: perform a dilution experiment. If the inhibition effect is purely competitive, diluting the reaction mixture will cause the inhibitor to dissociate more quickly because the substrate concentration becomes relatively larger. If the inhibition is non-competitive, dilution will relieve the effect proportionally to the reduction in inhibitor concentration, but the substrate concentration won't change the relief kinetics. This is an old-school method that doesn't require any fancy equipment. Just a steady hand and a stopwatch. I once used this approach to solve a six-month mystery involving a promiscuous inhibitor that was actually chelating a metal cofactor. That was non-competitive inhibition caused by sequestration, not by binding to the enzyme at all. You have to stay creative.
Why Mixed Inhibition Throws a Wrench in Everything
Let's be honest: pure competitive inhibition and pure non-competitive inhibition are idealized endpoints on a continuum. In the real world, you will frequently encounter mixed inhibition, where the inhibitor affects both the Km and the Vmax simultaneously. This happens when the inhibitor can bind to both the free enzyme and the enzyme-substrate complex, but with different affinities. The effect on the kinetic parameters is a hybrid. The Vmax goes down, and the Km might go up or down depending on whether the inhibitor prefers the free enzyme or the complex.
I've had senior scientists argue with me that mixed inhibition is just sloppy data. It's not. It's a legitimate biological phenomenon, especially in enzymes that undergo conformational changes during their catalytic cycle. When you're distinguishing competitive from non-competitive inhibition effects, you must always run a mixed inhibition model as a baseline comparison. If the mixed model fits significantly better than either pure model, you have a more complex situation. This usually means the inhibitor binding is influencing the enzyme's structural dynamics rather than just blocking a simple site.
For example, I studied a kinase inhibitor that showed a 3-fold increase in Km and a 20% drop in Vmax. That's not a clean case. The competitive model gave a reasonable fit, but the non-competitive model was terrible. However, the mixed model revealed that the inhibitor had a 10-fold higher affinity for the enzyme-substrate complex than for the free enzyme. That was crucial information for the medicinal chemistry team. It told them the inhibitor was essentially "substrate-trapping." If they had classified it as simple competitive inhibition, they would have pursued the wrong optimization strategy. Always check the residuals.
Real-World Pitfalls That Lead to Misclassification
Here are the top three mistakes I see in the literature and in peer review when it comes to distinguishing competitive from non-competitive inhibition effects. First, using too few data points. I've reviewed manuscripts where the authors used three substrate concentrations and called it a day. You cannot resolve a Km shift with three points. That's like trying to measure your height with a yardstick that has only two marks. You need density. Second, ignoring the solubility of the inhibitor. If your inhibitor precipitates at higher concentrations, you're not measuring inhibition; you're measuring light scattering. Run a dynamic light scattering control or a centrifugation assay. Third, assuming reversibility without proof. Some compounds form covalent adducts but look like reversible non-competitive inhibition on initial plots. If you don't run a dilution experiment or a dialysis recovery test, you will misclassify an irreversible inhibitor as reversible. That's a fatal error in drug discovery.
'Look—some inhibitors change their mode depending on the buffer. I've had compounds that were competitive in phosphate buffer and non-competitive in HEPES buffer. Why? Because the ionic strength affected the ionization state of a critical histidine residue in the active site. The inhibitor was actually binding to the deprotonated form of the enzyme. When the buffer shifted the pKa, the binding mode changed. This is rare, but it's real. If you're distinguishing competitive from non-competitive inhibition effects for a publication, always test at two different buffer conditions. It will protect you from embarrassing corrections later.
Why This Distinction Matters in the Real World
Let's zoom out. Why should you, the reader, care about this? Because the entire strategy for addressing an inhibition problem in a therapeutic or industrial context hinges on this diagnosis. If you have a competitive inhibition issue in a drug candidate, you can potentially solve it by increasing the dose or by designing a prodrug that elevates the local substrate concentration. Non-competitive inhibition is a harder problem. You need to change the chemical scaffold entirely or find an alternative pathway. I've seen entire development programs pivot based on a single mechanistic classification.
Consider the field of antimicrobial resistance. Many beta-lactamase inhibitors are competitive inhibition effects against the resistance enzymes. But the next-generation candidates are moving toward non-competitive mechanisms because they offer broader coverage and are less likely to be overcome by substrate overexpression. That's a direct consequence of understanding the kinetic difference. Similarly, in pesticide design, a non-competitive inhibitor of acetylcholinesterase is often more potent at lower concentrations than a competitive one, but it also carries a higher risk of off-target toxicity. The classification drives the risk assessment.
I once consulted for a biotech startup that was trying to inhibit a key metabolic enzyme in cancer cells. Their lead compound was showing a 50% reduction in Vmax with no change in Km. That screamed pure non-competitive inhibition. The CEO wanted to push it into animal trials immediately. I advised them to run a full reversibility assay first. Turns out the compound was forming a covalent bond with a cysteine residue. That's not reversible non-competitive inhibition; that's irreversible modification. The pharmacokinetic profile would be entirely different. They avoided a costly failure because we took the time to correctly distinguish competitive from non-competitive inhibition effects. Don't underestimate the financial implications of a misdiagnosis.
Common Questions About Distinguishing Competitive from Non-Competitive Inhibition Effects
Can a single inhibitor show both competitive and non-competitive behavior?
Yes, absolutely. This is called mixed inhibition. The inhibitor binds to both the free enzyme and the enzyme-substrate complex, but with different affinities. The Lineweaver-Burk lines will intersect somewhere between the x-axis and y-axis. You need to use a mixed model equation (including an alpha value) to accurately describe the behavior. Pure competitive or non-competitive are ideal cases; mixed is the reality for many allosteric inhibitors.
How do I tell if an inhibitor is competitive or non-competitive just from a single inhibition percentage?
You cannot. This is the most common rookie mistake. A single inhibition percentage at one substrate concentration tells you nothing about the mechanism. You need a full substrate-velocity curve at multiple inhibitor concentrations to see how the Km and Vmax are affected. If you only have one data point, you're guessing. Always design your experiment with at least five substrate levels and four inhibitor levels.
Is non-competitive inhibition always reversible?
No. This is a dangerous assumption. Some irreversible inhibitors (like those that form covalent bonds) can appear non-competitive in a standard assay because they reduce Vmax without changing Km. You must perform a dilution experiment or a dialysis step to confirm reversibility. If the activity does not recover after dilution or dialysis, you're dealing with irreversible inhibition, regardless of what the initial curve shape looks like.
Why does my non-competitive inhibitor sometimes shift the Km slightly?
That usually indicates you are dealing with mixed inhibition, not pure non-competitive inhibition. Pure non-competitive inhibition should leave the Km unchanged. A small shift in Km (less than 20%) could be experimental noise or a weak secondary binding event. If the shift is reproducible and larger than your standard error, fit your data to a mixed inhibition model. It's possible your inhibitor has a slight preference for binding to the free enzyme versus the complex.
Can I use an IC50 value to distinguish competitive from non-competitive inhibition?
Only if you measure the IC50 at multiple substrate concentrations. A competitive inhibitor will have an IC50 that increases as the substrate concentration increases. A non-competitive inhibitor will have an IC50 that stays constant regardless of the substrate level. If you measure IC50 at only one substrate concentration, you have no mechanistic information. Always run a Cheng-Prusoff correction if you want to convert IC50 to a true Ki value.